Concise total syntheses of (–)-jorunnamycin A and (–)-jorumycin enabled by asymmetric catalysis
Eric R. Welin, Aurapat Ngamnithiporn, Max Klatte, Guillaume Lapointe, Gerit M. Pototschnig, Martina S. J. McDermott, Dylan Conklin, Christopher D. Gilmore, Pamela M. Tadross, Christopher K. Haley, Kenji Negoro, Emil Glibstrup, Christian U. Grünanger, Kevin M. Allan, Scott C. Virgil, Dennis J. Slamon, Brian M. Stoltz
Science,
2019, 363, 6424, 270-275; DOI:10.1126/science.aav3421
01/2019
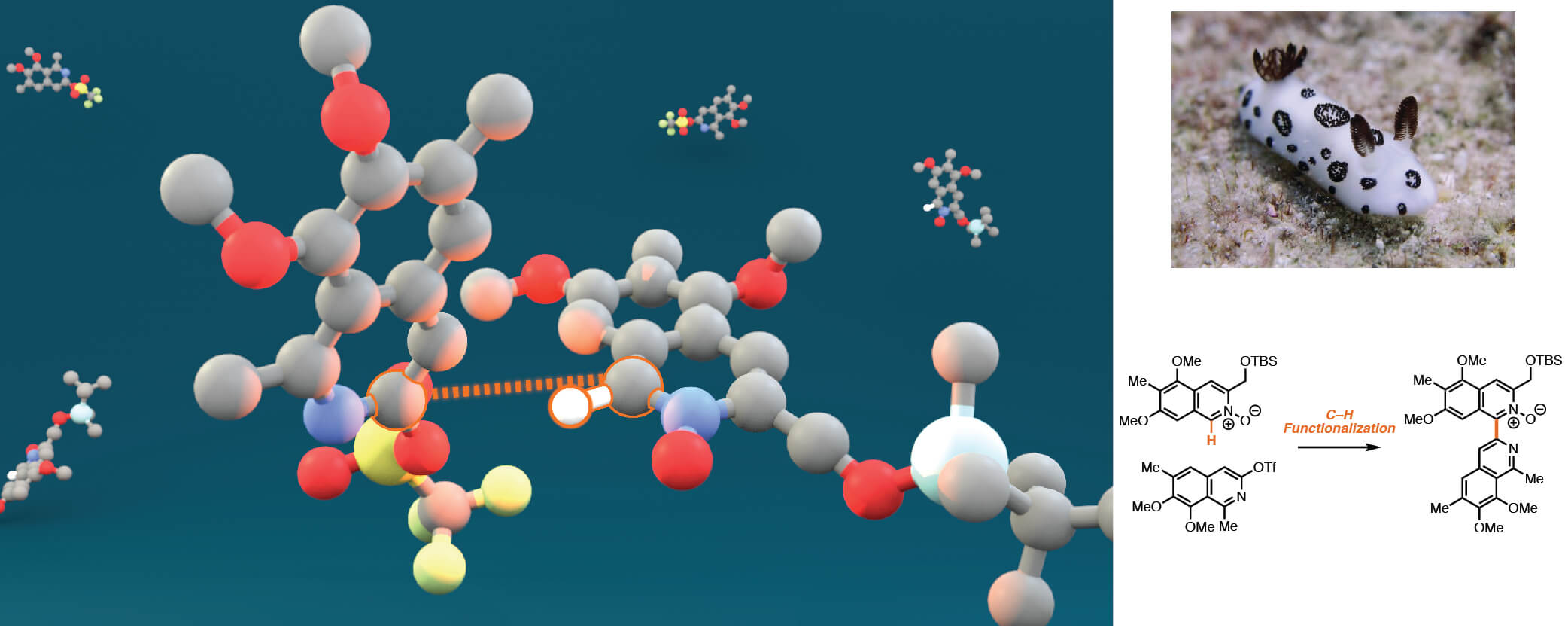
In this report in Science, the Stoltz Group, experts in the construction of complex molecules, report an approach that harnesses the potential of C–H functionalization to streamline the synthesis of a class of natural products that show significant anti-cancer and anti-biotic activity.
The strategy employs a palladium-catalyzed C–H activation to couple together two halves of the molecule, a modular approach that permits the synthesis of analogues for structure-activity relationship studies. Many discussions and meetings in the Center focused on this significant chemical transformation. A second key step in this synthesis is an enantioselective hydrogenation, during which four molecules of hydrogen are added to the molecule and a ring is formed in a single transformation.
Not only did this approach result in the shortest enantioselective total synthesis of two natural products (Jorunnamycin A; 15 steps and Jorumycin; 16 steps), but a library of analogues were prepared. In collaboration with the Slamon Group from UCLA it was found that two of the synthesized compounds demonstrate activity similar to that of known anticancer agents.